この講義で伝えたいこと
2012年3月26日からの試行期間を経て、2019年3月26日より、「患者副作用報告制度」が始動しました。副作用報告制度は医薬品のリスク管理のためにMRとして当然押さえておくべき知識であり、新たに追加された本制度を知らないことは、MRとしての意識を疑われると言っても過言ではありません。
この教材では、本制度の創設の経緯、制度の概要といった基本的な知識を押さえたうえで、これまでの報告状況をみていきます。本制度の意義を正しく理解し、医薬品リスク管理のための医薬品安全性監視活動につなげましょう。
患者副作用報告制度創設の経緯
まずは、患者副作用報告制度創設の経緯からみていきます。本制度の目的を理解しましょう。
これまでの副作用報告制度に加えられた「患者副作用報告制度」
2019年3月26日より、使用した医薬品の副作用かもしれないと感じた症状を患者自身がPMDAに直接報告できる「患者副作用報告制度」が本格始動しました。
医薬品医療機器等法に基いてこれまでに設けられている副作用報告制度として、製造販売業者が報告者である「企業報告制度」および「感染症定期報告制度」、医師・薬剤師などの医薬関係者が報告者である「医薬品・医療機器等安全性情報報告制度」があります。
医薬品医療機器等法に規定された副作用報告制度
| 制度 | 条項 | 報告者 | 報告対象製品 |
|---|---|---|---|
| 企業報告制度 | 第68条の10第1項 | 製造販売業者 | 医薬品・医薬部外品・化粧品・医療機器・再生医療等製品 |
| 感染症定期報告制度 | 第68条の24 | 製造販売業者 | 生物由来製品・再生医療等製品 |
| 医薬品・医療機器等安全性情報報告制度 | 第68条の10第2項 | 医薬関係者 | 医薬品・医療機器・再生医療等製品 |
編集部作成
ではなぜ、これらに加え、「患者副作用報告制度」が創設されたのでしょうか。
制度創設の背景にあった薬害事件
患者副作用報告制度創設の契機となったのは、薬害肝炎事件(※)です。
2008年、薬害肝炎事件の発生および被害拡大の経過、原因などの実態について多方面からの検証を行い、再発防止のための医薬品行政の見直しなどを提言することを目的に、厚生労働省に「薬害肝炎事件の検証及び再発防止のための医薬品行政のあり方検討委員会」が設置されました。そして2010年4月に、2年間の審議の取りまとめとして、「最終提言」が出されました。このなかで、薬害再発防止のための市販後安全対策として、患者からの副作用報告制度を創設すべきである、との提言がなされました。
※薬害肝炎事件:1971年から1990年頃に、フィブリノゲン製剤や血液凝固第Ⅸ因子製剤の投与により、同製剤に混入していたC型肝炎ウイルスに感染したとして、国および製薬企業に対し損害賠償を求める訴訟が2002年10月以降、全国5地裁で提起された。国はその責任を認め、2008年1月に和解内容を取り決めた「基本合意書」が、厚生労働大臣、原告団、弁護団の間で調印された。
「薬害再発防止のための医薬品行政等の見直しについて(最終提言)」より抜粋(赤字:編集部)
- 「患者からの副作用報告制度」(患者からの副作用に関する情報を活かせる仕組み)を創設すべきである。
- 患者からの副作用報告制度を創設し、この制度の下で得られる情報を安全対策に生かすとともに、おくすり相談で得られる情報も安全対策に活用すべきである。
そして、この最終提言を踏まえ、医薬品等の承認時や販売後の安全対策の強化、制度改正事項について調査審議するため、厚生科学審議会(※)下に設置された「医薬品等制度改正検討部会」での議論の結果、2012年1月に報告された「薬事法等制度改正についてのとりまとめ」においても、患者から直接副作用報告を得ることの有用性と、得られた副作用情報を安全対策に活用する必要性が指摘されました。
※厚生科学審議会:厚生労働省下に設置される審議会のひとつ。疾病の予防および治療に関する研究等、化学技術に関する重要事項を調査審議する。
患者自身が副作用の第一発見者となることもあり、患者から直接副作用報告を収集することも有用であると考えられる。患者からの副作用報告については、現在、厚生労働科学研究事業においてパイロットスタディが進められているが、これを推進し、得られた副作用情報を安全対策に活用すべきである。
「薬事法等制度改正についてのとりまとめ」より(赤字:編集部)
なぜ患者副作用報告が薬害再発防止策となるのか
アメリカ、イギリス、オランダなどの諸外国では、日本より早く、患者から直接副作用報告を受け付ける制度が導入されています。
たとえばアメリカでは、1993年より患者から副作用の疑い報告を受け付けています。EUでは、患者副作用報告制度を2012年7月までに各国の規制に取り込むことが指示され、EU加盟国で対応を行っています。
患者からの副作用報告の諸外国での導入状況
| 国 | 制度・システム・報告の仕組み |
|---|---|
| アメリカ | 患者からの副作用直接報告は、FDAのMedWatchの枠組みに含まれており、1993年に開始。医薬品安全性情報のポータルサイト「MedWatch」で、約130の関連団体や組織と連携し、FDAへの有害事象報告を受けるシステムを有している。 |
| イギリス | イエローカード副作用報告システム(Yellow Card Scheme)という独自の副作用報告システムがある。Yellow Card Scheme は、従来は医療従事者が使用するためのものであったが、2005年からは報告者を患者本人とその介護者に対しても開放している。 |
| オランダ | 国の独立機関である「オランダ薬剤監視センターLareb(Landelijke Registratie en Evaluatie van Bijwerkingen)」で2003年より患者からの副作用報告の受け付けを開始している。 |
| ドイツ | 医師、薬剤師、患者、被害患者の弁護士から、製薬メーカーや医師会・歯科医師会・薬剤師会の医薬品委員会を経由し、もしくは直接にBfArMが情報を収集し、BfArMからEMA・WHOや各情報源に対して情報提供している。 |
編集部作成(参考:平成25年度PMDA委託 医薬品・医療機器等の安全性情報の入手・伝達・活用状況等についての調査)
ではなぜ、患者から直接副作用報告を受け付けることが、薬害再発防止につながるのでしょうか。
PMDAは、多様な観点からたくさんの副作用報告を集めることで未知の副作用が起こっていることを認識できると考えています。
医薬品の副作用報告は、医薬品の安全対策を行う上で非常に重要な情報のひとつです。たくさんの副作用報告をいただくことで、それまで知られていなかった副作用が起こっていることを認識し、医療関係者や患者さんに対して注意喚起をすることができます。こうした活動が行われることで、より安全に医薬品をご使用いただけるものと考えています。
医薬品の安全対策において、多様な観点からの報告を活用することは有用であると考えられることから、患者さんやご家族から直接副作用報告を集めています。
PMDAウェブサイト「患者副作用報告に関するQ&A」より(赤字:編集部)
患者副作用報告では、報告された症状の原因として適切とは考えにくい薬剤が特定される場合があるといったデメリットも考えられますが、医療者による選別を経ずに患者の体験が患者の言葉で報告されるため、これまで見落とされていたかもしれない副作用を収集できたり、早期発見につながる可能性が高まるというメリットがあります。
患者副作用報告のメリットとデメリットの例
<メリット>
- 患者は医療従事者と比較して既存の製品情報で大きく扱われていない新規の副作用を報告する。
- 医療従事者の選別や解釈を経ずに副作用が報告される。
<デメリット>
- 報告された症状の原因として適切とは考えにくい薬剤が特定される場合がある。
- 患者が副作用を疾患の症状と勘違いして報告する可能性がある。
編集部作成(参考:「欧米における患者と医薬品情報―患者からの副作用報告」など)
患者副作用報告制度の概要
ここからは患者副作用報告制度の概要についてみていきます。本制度の基本的な知識を押さえましょう。
制度の概要を押さえよう
患者副作用報告制度は、医薬品による副作用が疑われる症例についての情報を、患者またはその家族から直接収集することにより、医薬品の安全対策に活用することを目的としています。
本制度は、これまで副作用報告を行っていた製造販売業者や医薬関係者とは異なる視点からの副作用情報を収集することが狙いです。そのため、報告者は、医薬品の使用により副作用が疑われる症状が現れた患者自身、またはその家族です。
医師から処方された医療用医薬品だけでなく、ドラッグストアなどで購入した一般用医薬品・要指導医薬品(※)による副作用、予防接種ワクチンによる副反応についても報告の対象となります。
※要指導医薬品:2013年の薬事法(当時)改正により新設された医薬品の区分。医療用医薬品からOTC医薬品にスイッチされて間がない医薬品。適正使用のため、薬剤師が直接対面で文書による情報提供が必要。
「患者副作用報告制度」の概要
- 目的
医薬品による副作用が疑われる症例についての情報を、患者またはその家族から直接収集することにより、医薬品の安全対策に活用する - 報告者
医薬品の使用により副作用が疑われる症状が現れた方またはその家族 - 報告対象医薬品
国内で製造販売されている医療用医薬品、要指導医薬品、一般用医薬品
「『患者からの医薬品副作用報告』実施要領」 より編集部作成
なお、医薬品の副作用であると思われた症状であれば、どのような症状でも、症状の程度にかかわらず報告することができます。医療機関において医薬品の副作用であると診断されていない症状についても報告可能であり、医療機関に報告の了解を得る必要もありません。
報告は、PMDAのウェブサイトから、または郵送で行います。
ウェブサイトからの報告は、PMDAウェブサイトホームページ右下の「患者さんからの医薬品副作用報告はこちらから」のバナーをクリックし、「患者の皆様からの医薬品副作用報告」のページにアクセスします。
PMDAウェブサイトからの報告の入り口
PMDAウェブサイト より作成
郵送による報告は、同じくPMDAウェブサイトから同ページにアクセスし、「郵送による報告」を選択します。「郵送による患者副作用報告」ページから、報告様式をダウンロードして必要事項を記入のうえ、PMDAへ郵送します。報告様式はこの方法で入手するほか、電話で報告様式を請求し、郵送してもらうことも可能です。
こうした対応により、インターネット環境が整っていない患者さんや家族も報告できるよう配慮されています。
ウェブ上のフォームへ入力、または様式へ記入していくことで、報告者に関する情報、患者に関する情報、副作用が疑われる症状を引き起こしたと思われる医薬品に関する情報、他に使用していた医薬品に関する情報、症状に関する情報、詳しい情報を聞くことができる医療機関に関する情報について報告します。
フォームでは、副作用を引き起こしたと思われる医薬品については最大5つ、副作用の症状については10個まで入力することができます。
【参考】PMDAウェブサイト掲載入力項目
患者副作用報告での報告項目
- 報告者に関する情報
- 患者に関する情報
- 副作用が疑われる症状を引き起こしたと思われる医薬品に関する情報
- 他に使用していた医薬品に関する情報
- 症状に関する情報
- 詳しい情報を聞くことができる医療機関に関する情報
「『患者からの医薬品副作用報告』実施要領」 より編集部作成
報告した情報がどのように安全対策にいかされるか
下の図は、患者または家族から副作用報告がなされたその後の流れを示したものです。
患者副作用報告後の流れ
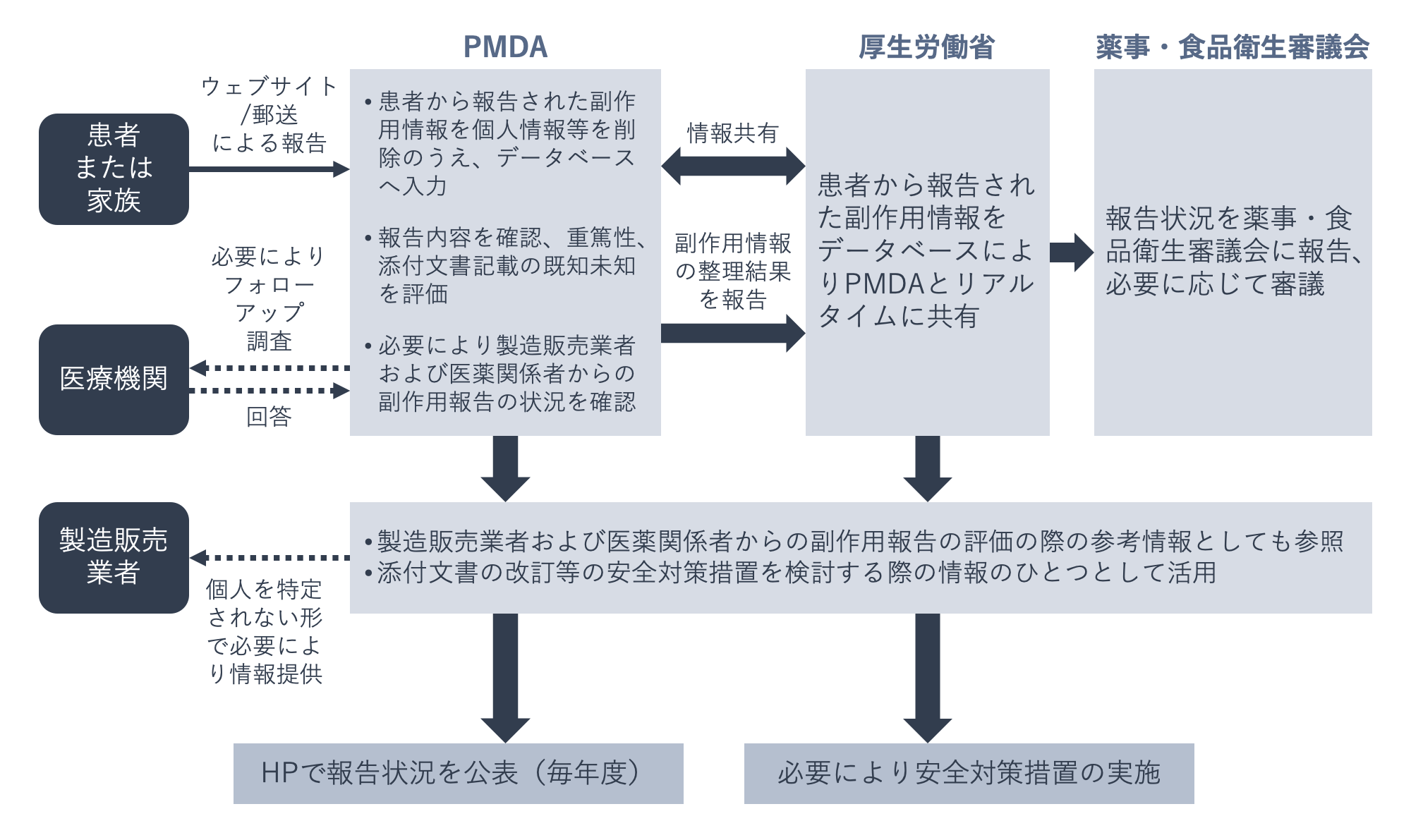
平成30年度第3回薬事・食品衛生審議会医薬品等安全対策部会資料 より作成
報告された情報についてPMDAでは、症状の重さや添付文書において既に知られている副作用であるかどうかの確認と、製造販売業者・医療機関から報告された副作用等の情報も含め、さらなる安全対策が必要かどうかの検討を行います。
副作用情報の分析・評価を行うためにさらに詳細な情報が必要と判断した場合、PMDAが医療機関に対してフォローアップ調査を行います。
報告された情報は、個人情報を除いて、一定期間ごとに厚生労働省の薬事・食品衛生審議会医薬品等安全対策部会にて報告されます。さらに、個人が特定されない形に加工したうえでPMDAウェブサイトで公表されるほか、安全対策の一環として、当該医薬品を供給する製造販売業者に提供されることがあります。
このように、報告データは、副作用の発生動向の把握や安全対策措置の検討に役立てられます。