
編集協力:A・T
現役の某外資系製薬メーカーの研究職でアメリカに籍を置く。日本法人では臨床開発部でオンコロジーを担当。某内資系製薬メーカーでMR職を15年経験。全国の大学病院と国公立病院で人脈を作り、転職先の外資系製薬メーカでは営業統括職となる。その後、研究部署へ異動。現在は主にオンコロジーのゲノム研究に携わっている。
私はMR出身の臨床開発職ですが、大学病院と国公立の病院、さらに診療所も担当していました。今から30年前なのでまだプロモーションが厳しい時代ではありませんでした。しかし大学病院の教授や大病院の部長クラスの先生と頻繁に面談するMRは少なかったです。これは現在でも変わりません。私は医師のポジションに関係なく、医師としての考え方、何が研究したいのか、趣味は何か、家族構成など、すべてコミュニケーションをとって教えてもらいました。さらに先生の専門分野の専門知識をご教授いただいたりもしました。そこから別の先生を紹介していただくこともありました。
この経験が臨床開発職に異動した今も、宝となっています。MR時代にお世話になった先生方がいま、開発にあたっている新薬の領域で地域のオピニオンリーダーである先生を紹介してくださり、直接お話させていただけるからです。MR時代に人間関係を築けた先生方には、直接電話をすることもできるし、面談もできます(現在は新型コロナの影響でリアルでの対面は控えていますが)。
もし今、部長クラスの先生と面談したいのに勇気が湧かないMRは、思い切ってドアをノックしましょう。叱られることは無料です。私もこれまで何度も、叱られてきました。しかし、そこからはじまるご縁もあります。熱意と誠意をもって仕事をするMRを応援してくれる先生は必ずいます。
この講義で伝えたいこと
医療の世界は日進月歩。創薬の技術も日々、進歩しています。しかし、創薬技術がいかに優れたものであったとしても、生まれた医薬品がすでに完璧な状態であるということはありません。
臨床の現場に送り出された医薬品はまだ製造販売承認を取得するための治験データを蓄積し終えただけで、言うなれば「仮免許」の状態です。医薬品の効果の反面である副作用など、未知の可能性を含んだまま、患者さんの体に入っていくのです。販売後に新たな用法や別の疾患への有効性が発見されることもあります。医薬品がより適正に、より効果的に使われるよう、しっかりと育てていかなければいけません。
製薬企業の努力だけでは医薬品を育てることはできません。患者さんの声、そして医療者からの情報収集が必要不可欠です。そこで重要になってくるのが、製造販売後調査です。製造販売後調査に関わるGPSP省令を理解し、MRとして自社医薬品の有効性と安全性の確保のために情報提供・収集・伝達活動ができているか、この機会に見つめ直してみましょう。
本編の前に
本編をはじめる前に、独自に行ったアンケート調査の結果の紹介と、基本的な用語の確認をしておきます。
一般の方へのアンケート
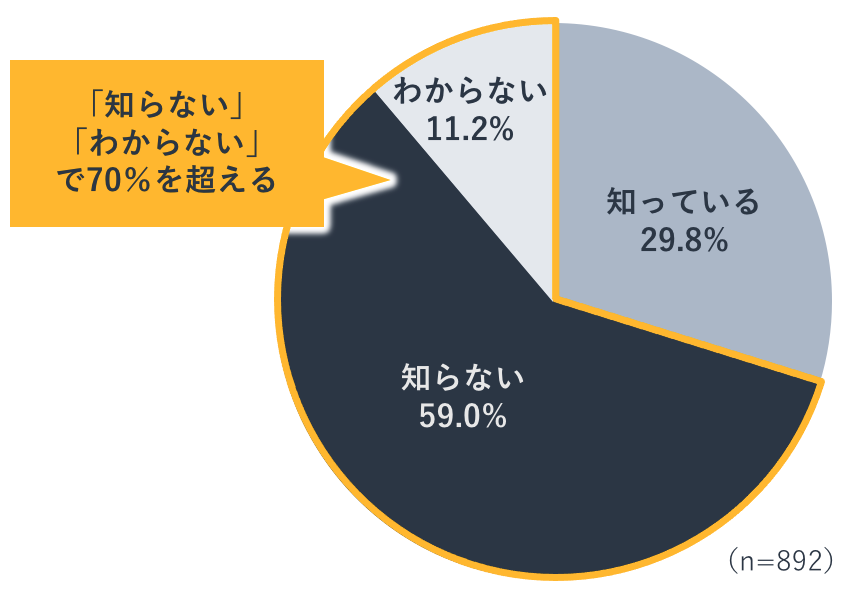
医薬品の製造販売に関連する仕事に従事している者であれば、市場に出た医薬品(新医薬品)が再度、その有効性と安全性について審査を受ける制度があることを当然知っているでしょう。ではこの制度についての一般の方の認知度はどれぐらいだと思いますか?
医療従事者と医薬品の製造販売に関する仕事に従事する人を除く20歳以上の男女を対象に年代・性別の偏りなくアンケート調査を行ったところ、その制度を「知らない」と回答した人の割合は59%にのぼりました(n=892)。「わからない」と回答した人も合わせると、70%を超えます。
医薬品は、承認を受けて販売されてから一定期間の後に、
その有効性と安全性を再確認するために、再度、審査を受けていることを知っていますか?
編集部作成
この結果から、一般の方には医薬品が厳重な安全対策を施されて患者さんのもとに届けられていることがほとんど浸透していない、と言えます。
「GPSP省令」とは
突然ですが、ここでクイズです。
MRの業務に欠かせない「GPSP省令」とは、何についての省令か、正しく答えられますか?
正解は、「医薬品の製造販売後の調査及び試験の実施の基準に関する省令」です。
GLP、GCP、GMP、GQP、GVP…。
GPSP省令のほか、医薬品の製造販売に関係する規制は似たような名称でいくつもあり、MR認定試験の際に必死で詰め込んだけれど、今となってはどれが何の基準であるか、あやふやになっている方も多いように思います。
単なる暗記ではすぐに忘れてしまいます。忘れないコツは、きちんと理解することです。「GPSP」とはどんな英語の略称であるかを理解することで、忘れなくなります。
「GPSP」は「Good Post-Marketing Study Practice」の略です。これを単語に分解して意味を考えてみましょう。
Goodはもちろん「良い」ということですが、日本語での「確実な」や「適した」といった意味にもなります。Post-Marketingは市場後、Studyは調査・試験、Practiceは実践、です。続けると、確実な(適した)市場後の調査・試験の実践。つまり、「医薬品の製造販売後の調査及び試験の実施の基準」ということになります。
「GPSP省令」とは、何についての省令か?

編集部作成
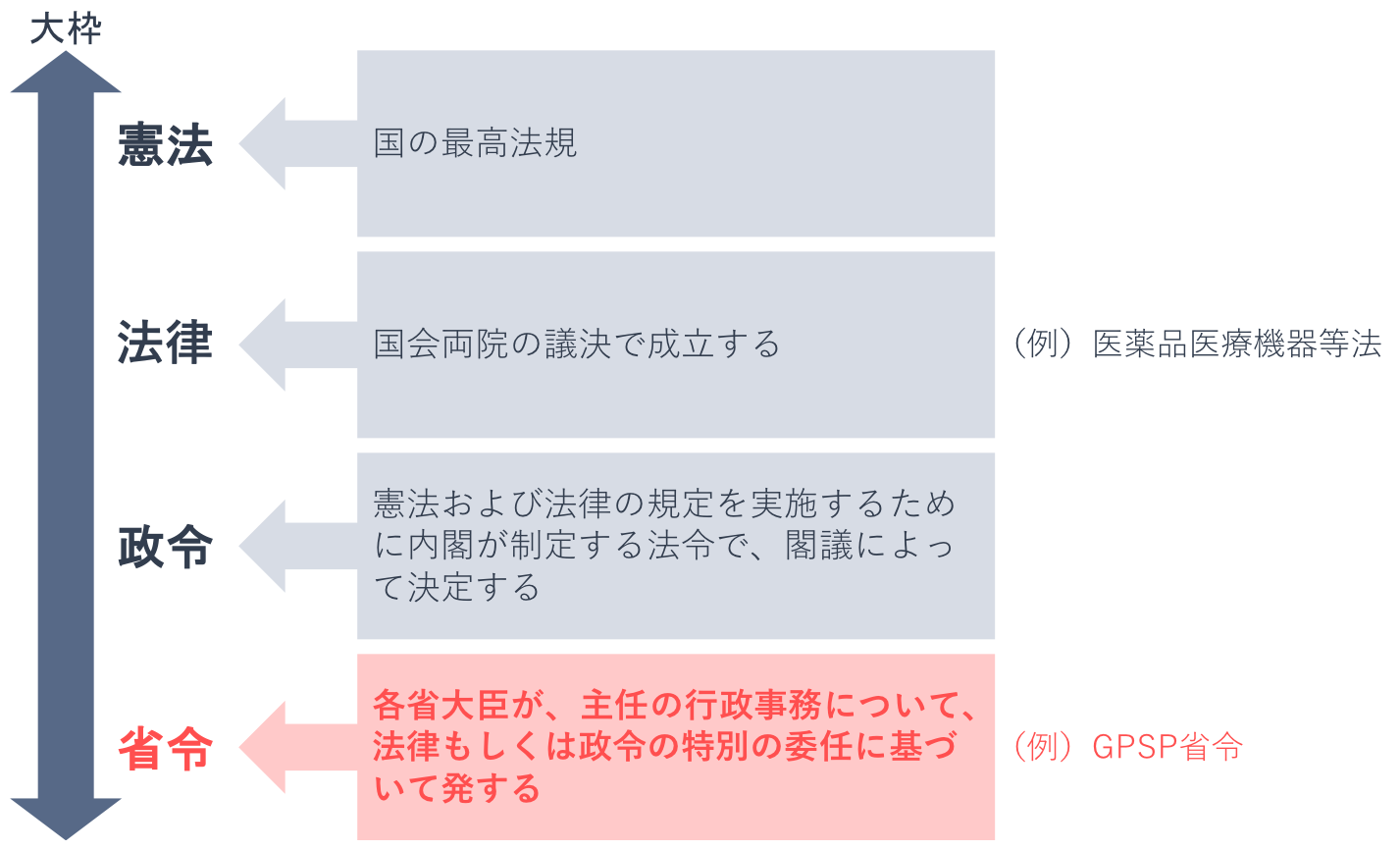
ちなみに省令とは、各省庁の大臣が定める命令です。法律や政令で規定していない細かなことを定めており、法律を補足するものと理解するとよいでしょう。
GPSP省令は厚生労働省令であり、薬事関連法規の根幹をなす法律である「医薬品医療機器等法」(※)を補足するものです。
※医薬品医療機器等法:正式名称は「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律」。薬機法とも呼ばれる。
日本の法体系
医薬品食品衛生研究所資料より改変作成
GPSP基本的知識のおさらい
では、ここから、講義の本編をはじます。
まずは、GPSPの基本的な知識をおさらいしておきましょう。
GPSPは医薬品の再審査と再評価に関わる
第一条 この省令は、医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律第十四条の四第五項及び第十四条の六第四項の厚生労働省令で定める基準のうち製造販売後の調査及び試験に係るもの及び医薬品の製造販売業者又は外国製造医薬品等特例承認取得者が医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律施行規則第十四条第一項に規定する医療用医薬品について行う製造販売後の調査及び試験の業務に関して遵守すべき事項を定めるものとする。
「医薬品の製造販売後の調査及び試験の実施の基準に関する省令」より
GPSP省令の第一条にはこの省令の趣旨が書かれています。要約すると、「この省令は、医薬品医療機器等法の14条の4第5項、14条の6第4項の、製造販売後の調査と試験の業務に関して遵守すべき事項を定めるものである」となります。
医薬品医療機器等法の14条の4は新医薬品等の再審査について、14条の6は医薬品の再評価について規定するものです。つまりGPSP省令は、医薬品の再審査と再評価のための製造販売後調査・試験の基準を定めたものということです。
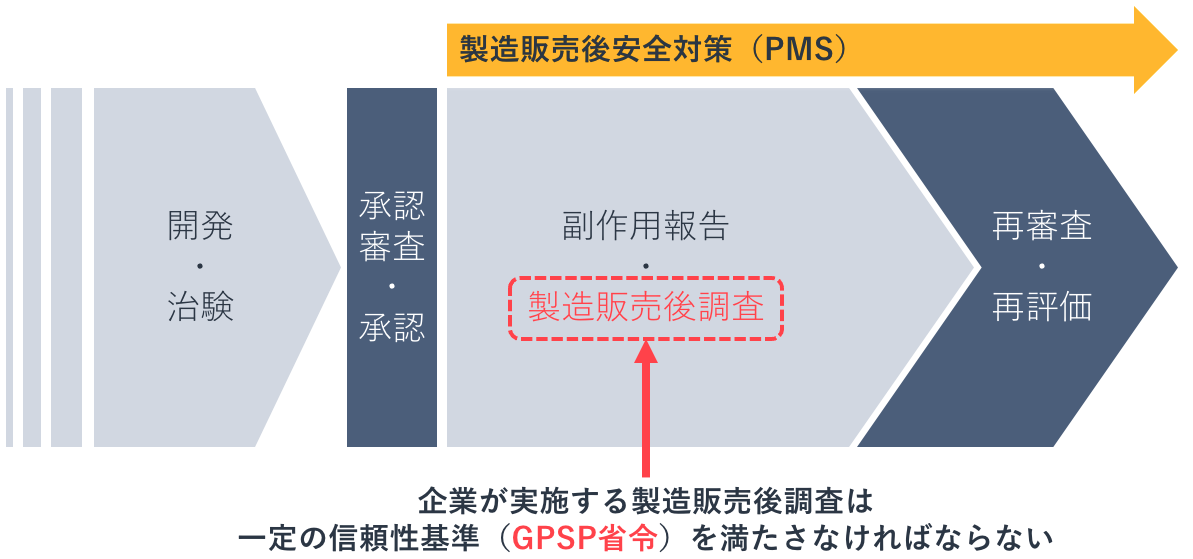
ある医薬品が製造販売承認を受けた時点での有効性・安全性の評価は、患者数や患者背景(併用薬、年齢など)が限定された状況下での、限られた情報によるものです。市販後は使用患者数が急増し患者背景が多様化するため、承認時に判明しなかった副作用が顕在化することがあります。そのため、市販後の安全対策(PMS:Post-Marketing Surveillance)が非常に重要になります。
PMSには、3つの柱と呼ばれる副作用報告・再審査・再評価という制度があります。その再審査と再評価に関わってくる製造販売後の調査は適正に実施されなければならず、信頼性が確保されたものでなければなりません。有効性・安全性の調査項目や調査方法が、実施する企業ごとにバラバラであっては客観的な評価ができなくなります。そのため、企業が実施する製造販売後調査は一定の信頼性基準を満たさなければならず、その基準を定めるのがGPSP省令です。
医薬品の製造販売後の安全対策
厚生労働省資料より改変作成
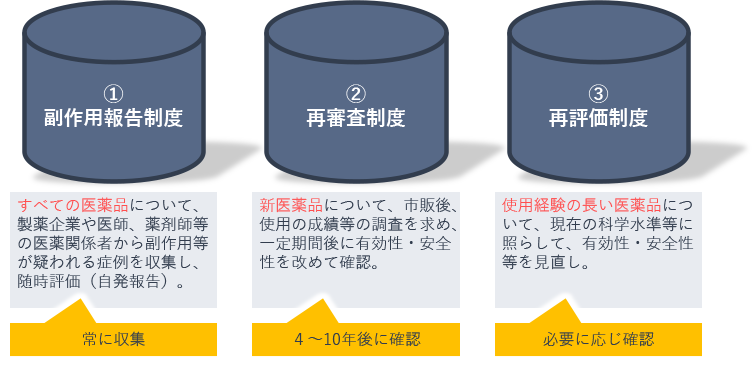
PMSの3つの柱と呼ばれる副作用報告制度・再審査制度・再評価制度についても、簡単におさらいしておきます。
PMSの3つの基本的な柱
厚生労働省資料より改変作成
副作用報告制度は、すべての医薬品について、常に収集し随時評価するものです。再審査制度は、新医薬品について、市販後、一定期間後に有効性と安全性を改めて審査するものです。再評価制度は、使用経験の長い医薬品について、必要に応じて有効性と安全性を見直すものです。
【参考】これらとは別に、品質再評価という制度もある。これは後発医薬品の品質を確認するためのもので、1995年3月までに承認申請された医療用医薬品の内用固形製剤について溶出試験を行う。品質が確認されたものはその後『医療用医薬品品質情報集(日本版オレンジブック)』に掲載されることになる。
製造販売後調査の整理
製造販売後調査に話を戻します。
製造販売後調査には、「使用成績調査」、使用成績調査の一類型として「特定使用成績調査」、「製造販売後臨床試験」が規定されていましたが、2017年10月に改正GPSP省令が公布され(2018年4月1日施行)、これまでの「使用成績調査」、「製造販売後臨床試験」に、新たに「製造販売後データベース調査」を加えた計3種類の調査・試験が規定されました。
そして、「使用成績調査」の分類の整理が行われました。これまで狭義の意味で使われてきた、患者の条件を定めず実施する使用成績調査が「一般使用成績調査」と名称づけられ、特定の医薬品を使用する者の情報と使用しない者の情報との比較評価が新たに「使用成績比較調査」と規定されました。
「特定使用成績調査」はこれまでと同様、使用成績調査のひとつに位置づけられています。
製造販売後調査の体系
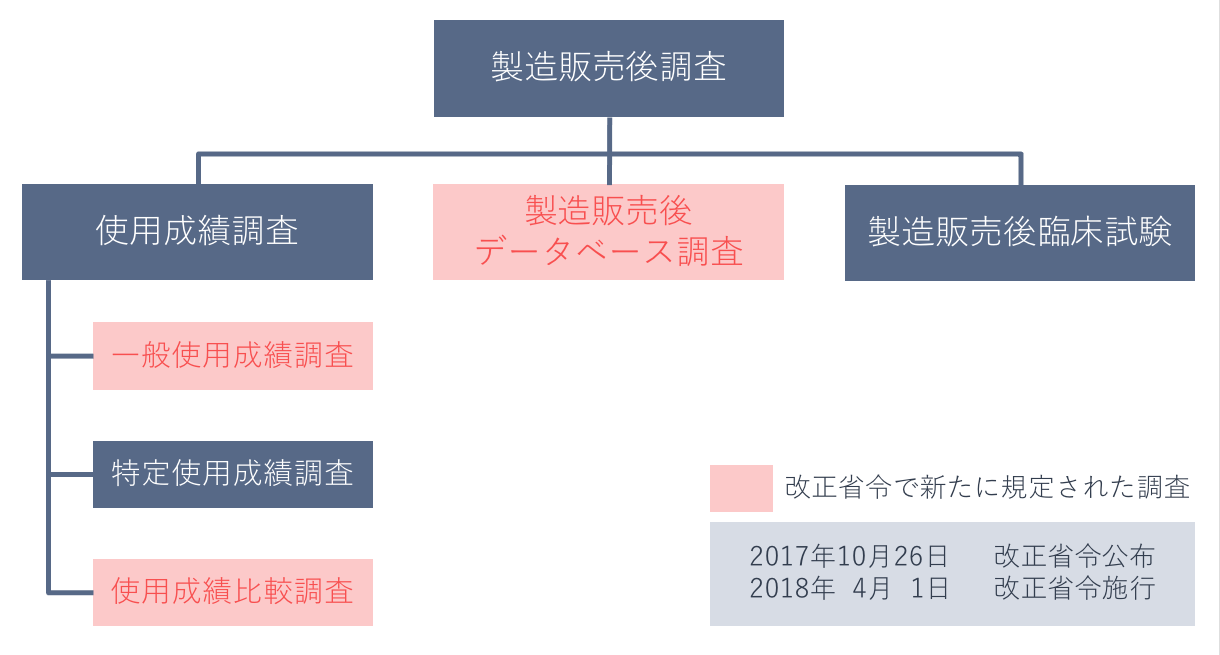
厚生労働省資料より作成
表は、それぞれの調査・試験の定義を詳しく示したものです。
各調査・試験の定義
| 区分 | 細分類 | 定義 |
|---|---|---|
| 使用成績調査 | – | 医療機関から収集した情報を用いて、診療において、医薬品の副作用による疾病などの種類別の発現状況並びに品質、有効性および安全性に関する情報の検出または確認のために行う調査であって、以下細分類に掲げるもの |
| 一般使用成績調査 | 医薬品を使用する者の条件を定めることなく行う調査(使用成績比較調査を除く) | |
| 特定使用成績調査 | 小児、高齢者、妊産婦、腎機能障害または肝機能障害を有する者、医薬品を長期に使用する者その他医薬品を使用する者の条件を定めて行う調査(使用成績比較調査を除く) | |
| 使用成績比較調査 | 特定の医薬品を使用する者の情報と当該医薬品を使用しない者の情報とを比較することによって行う調査 | |
| 製造販売後 データベース調査 |
– | 医療情報データベースを用い、医薬品の副作用による疾病などの種類別の発現状況並びに品質、有効性および安全性に関する情報の検出または確認のために行う調査 |
| 製造販売後臨床試験 | – | 治験、使用成績調査、製造販売後データベース調査の成績に関する検討を行った結果得られた推定などを検証し、または診療においては得られない品質、有効性、安全性に関する情報を収集するため、承認された用法、用量、効能・効果に従い行う試験 |
「医薬品の製造販売後の調査及び試験の実施の基準に関する省令」より編集部作成
MRの皆さんには、長らく「使用成績調査」と呼んできた「一般使用成績調査」と、小児や高齢者、妊産婦、腎機能障害または肝機能障害を有する患者、医薬品を長期に使用する患者など、使用条件が定められた患者について行う「特定使用成績調査」は馴染み深いものでしょう。
「使用成績比較調査」は、これまでも使用成績調査のひとつとして実施されてきたものですが、特定の医薬品を使用する患者の情報だけでなく、その医薬品を使用していない患者の情報についても医療機関から収集して比較を行うという調査が可能であることを明確にするため、省令改正により新たに規定されました。
「一般使用成績調査」は、特定使用成績調査と使用成績比較調査以外の使用成績調査が該当し、これら使用成績調査は、医療機関から直接収集する情報を用いる調査です。
対して、新たに規定された「製造販売後データベース調査」は、「医療情報データベース」を用いて、副作用の発現状況や、医薬品の有効性・安全性などに関する情報の検出、確認のために行う調査です。
それぞれの調査・試験の内容を整理して、しっかり把握しておきましょう。
製造販売後データベース調査とリアルワールドデータ
次に、製造販売後データベース調査とリアルワールドデータの関係をみていきます。
「医療情報データベース」とは
先ほど、「製造販売後データベース調査」は「医療情報データベース」を用いて行う調査、と説明しました。では、「医療情報データベース」とは、いったい何でしょうか。
「医療情報データベース」とは、一定の期間において収集される診療録その他の診療に関する記録、診療報酬請求書、疾病登録等に関する情報の集合物であって、それらの情報を電子計算機を用いて検索することができるように体系的に構成したものをいう。
「医薬品の製造販売後の調査及び試験の実施の基準に関する省令」より
GPSP省令では「医療情報データベース」を、診療録や診療報酬請求書、疾病登録(※)などに関する情報の集合物で、電子計算機を用いて検索することができるように体系的に構成したもの、と定義しています。
※疾病登録:ある疾病が発生した場合、それを診察した医師から衛生関係当局や自治体への患者の届出に基づいて、登録が行われる。代表的なものに全国がん登録、脳卒中登録がある。(一般社団法人日本疫学会ウェブサイトより引用)
具体的には、病院情報システムデータ(電子カルテデータ、診断群分類別包括評価(DPC )データ等)、診療報酬及び調剤報酬明細書(健康保険組合レセプトデータ等)、疾患登録データ等の電子的な医療情報(以下「医療データ」という。)を体系的に集積したデータベースが想定される。
薬生薬審発0221第1号 厚生労働省医薬・生活衛生局医薬品審査管理課長通知 より
厚生労働省通知ではこれを具体的に、電子カルテデータやDPCデータなどの病院情報システムデータ、診療報酬・調剤報酬明細書といったレセプトデータ、疾患登録データなどの医療データを体系的に集積したデータベース、としています。
つまり、臨床試験などで構築するデータとは異なり、実臨床の環境で収集された各種データを集積したデータベースを指します。MRの皆さんは、こうしたデータを指す「リアルワールドデータ」や、「リアルワールドエビデンス」といった言葉のほうが、耳にする機会が多いかもしれません。
リアルワールドデータの利用がなぜ必要とされたのか
従来の市販後安全対策は医療機関や製薬企業からの副作用自発報告や使用成績調査を主な情報源として行われてきましたが、課題が存在します。
たとえば副作用報告には、臨床の現場で副作用と認識された事象の一部しか報告されないといった報告バイアスを受ける、対象医薬品の使用者数(母数)が不明なため副作用の発生頻度を評価できない、原疾患による症状との判別ができない、併用薬が多数存在する場合には他剤とのリスクの比較ができない、といった課題があります。
また使用成績調査では、通常、対象群を設定せず、調査対象とする医薬品に曝露した症例についてのみ情報を収集するため、副作用の発生頻度は推定できますが、相対的なリスクを定量的に評価することはできないという課題があります。
ほかにも、安全対策の措置をとった前後での副作用発生頻度の比較ができず、安全対策措置が副作用低減に効果があったか検証できないという課題もあります。
従来の安全対策の課題
- 医薬関係者が報告しなければ、副作用の存在がわからない(報告バイアスを受ける)
- その医薬品を投与されている人数を把握できない(分母が不明のため副作用発生頻度が不明)
- 原疾患による症状と「副作用」の鑑別が難しい
- 他剤との副作用発生頻度の比較ができない
- 安全対策措置前後での副作用発生頻度の比較などができない
厚生労働省資料より改変作成
これらの課題を解決するため、より多様な情報源と新たな安全性評価の方法が求められ、医療情報データベース、つまりリアルワールドデータの利用が必要とされたのです。
現状、日本では、リアルワールドデータは大きく2種類に分けられます。ひとつめがPMDAの展開するMID-NET(※)や厚生労働省所管のNDB(※)などの医療記録データ、もうひとつが「疾患レジストリ」や「患者レジストリ」とも呼ばれる疾患(疾病)登録システム、別の言葉に置き換えると患者コホート研究とも言えるものです。
※MID-NET:協力医療機関でのオーダリング、検査結果などを含む電子カルテデータやレセプトデータ、DPCデータなどの電子診療情報をデータベース化し、それらを解析するシステム。
※NDB(National Database):厚生労働省の管轄でレセプト情報および特定健診情報を格納する国内最大のデータベース。
2種類のリアルワールドデータ

厚生労働省資料より作成
レジストリとは、ある特定の情報の収集を目的に作成されるデータベースで、収集する情報の種類で分けると、疾患レジストリ(患者レジストリ)のほか、曝露レジストリや診療行為レジストリなどがあります。国内の疾患レジストリの例として、がん、希少がん、筋ジストロフィー、筋萎縮性側索硬化症(ALS)、糖尿病など、多数のレジストリがあります。
リアルワールドデータは日常診療の現場から得られるデータのため、他の集団にも一般化しやすい(外的妥当性が高い)といった長所や、患者あたりのコストが安いという長所があります。しかし一方で、二次利用を前提に収集されているデータではないために、実施したい分析に必要な項目が収集されていなかったり、収集されていても欠損していたり、データが標準化されていなかったりといった、完全性・正確性が低いという短所があります。
比較して、臨床試験で得られるデータは、厳格に定められた基準のもとで目的の情報を収集するため、完全性・正確性が高く、内的妥当性の高いデータです。
リアルワールドデータの長所と短所
| 長所 | 短所 |
|---|---|
|
|
編集部作成
このようにデータの特性や長所と短所を考慮して、リアルワールドデータの利用に適した調査か否かを判断しなければなりません。
厚生労働省は、リアルワールドデータを活用することで可能になる安全対策として、たとえば、同種同効薬との副作用発現頻度を比較することができる、調査対象医薬品使用群と非使用群での有害事象の発生頻度を比較することで、有害事象が疾患自体の症状によるものか判別が可能になる、安全対策措置実施前後の副作用発現割合を比較することで、安全対策措置が副作用低減に効果があったかを検証できる、などを想定しています。
医療情報の活用により可能となる安全対策の例
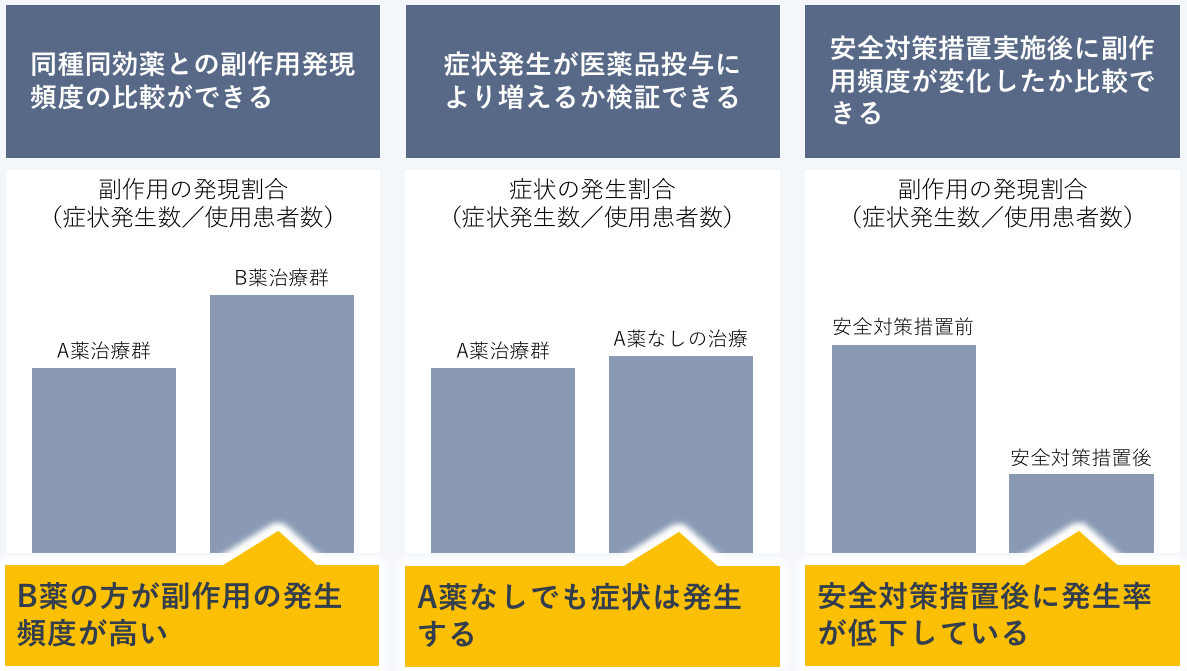
厚生労働省資料より改変作成
実際にMID-NETを活用した分析として、新規の抗血栓薬プラザキサによる出血の副作用と、古典的薬剤のワーファリンによる出血の発現頻度を比較して、プラザキサの出血リスクはワーファリンより高くないことを解析した例などを開示しています。
MID-NETの活用例:他剤との副作用頻度比較
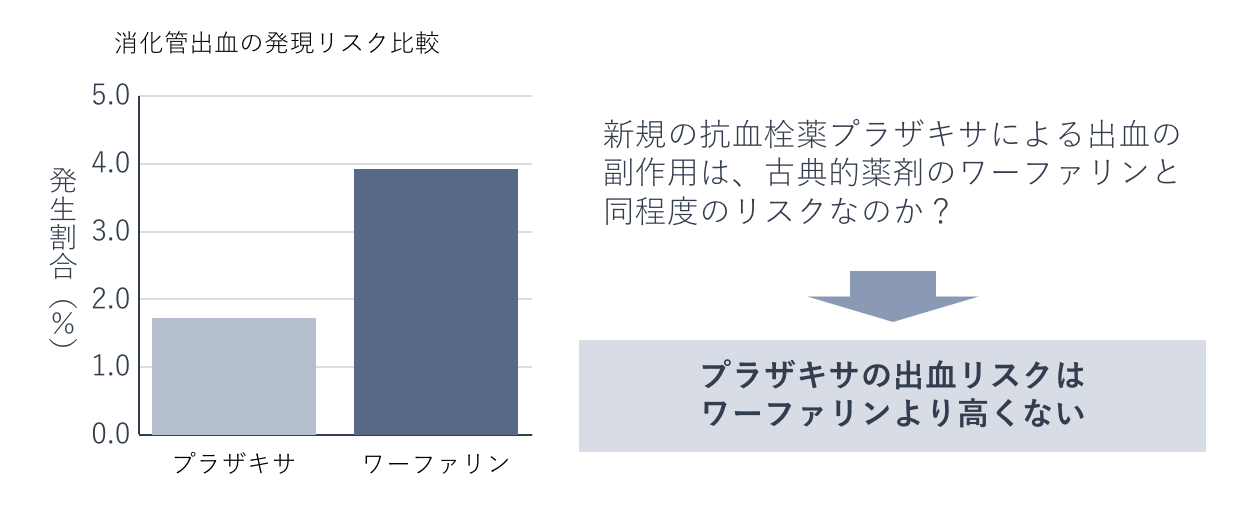
厚生労働省資料より作成
規制判断に活用され始めたリアルワールドデータ
2018年4月より本格運用を開始したMID-NETは、現在は500万人以上のデータが解析可能になっているとされており、製造販売後調査などの医薬品の安全対策と、公益性の高い調査・研究を目的に、行政・民間企業・アカデミアでの二次的利用が認められています。
現在までに製薬企業が製造販売後調査にMID-NETを活用した事例として、表に示した4件(4品目)があります。
製造販売後調査にMID-NETを活用した事例
| 企業名 | 調査・研究の内容 |
|---|---|
| ファイザー株式会社 | イブランスカプセルの再審査申請に係る安全性検討事項の調査 |
| 第一三共株式会社 | 「プラリア皮下注60mgシリンジ」の再審査申請に係る安全性検討事項の調査 |
| MSD株式会社 | アトーゼット配合錠LDおよびアトーゼット配合錠HDの再審査申請に係る安全性検討事項の調査 |
| 第一三共株式会社 | ミネブロ錠1.25mg/ミネブロ錠2.5mg/ミネブロ錠5mgの再審査申請に係る安全性検討事項の調査 |
PMDAウェブサイトより作成
諸外国においても、疾患レジストリなどのリアルワールドデータに関するデータベース整備が進んでおり、国際的な動向として、医薬品の承認といった規制判断へリアルワールドデータを導入する向きがあります。
規制判断へのリアルワールドデータ活用が世界で最も進んでいる国のひとつが、アメリカです。アメリカでは、医療製品の開発と臨床現場への導入を加速させることを目的に、2016年に21st Century Cures Act(21世紀治療法)が成立し、リアルワールドデータの活用が法律に明文化されました。
規制判断への活用事例をひとつ紹介します。2019年4月、FDA(※)は乳がんの分子標的薬パルボシクリブについて、リアルワールドデータのみに基づき、男性乳がんへの適応拡大を承認しました。
※FDA(Food and Drug Administration):医薬品の認可や違反取締を行うアメリカの政府機関。日本では「アメリカ食品医薬品局」と呼ばれることもある。
アメリカで規制判断にリアルワールドデータを活用した事例
パルボシクリブの適応拡大
- パルボシクリブはER陽性HER2陰性、閉経後の進行乳がんに対するCDK4/6 阻害薬
- 男性乳がんは全乳がん患者の1%未満、大部分はER陽性HER2陰性
- IQVIA claims data ・Flatiron Health EHR data・Pfizer global safety DBの3つを用いてパルボシクリブ有と無の患者間で、奏効率、治療継続期間、安全性を比較
2019年4月、FDAはリアルワールドデータのみに基づき、
男性乳がんへの適応拡大を承認
抗悪性腫瘍薬開発フォーラム資料より作成
規制当局がリアルワールドデータ活用を進める動きは、日本でも始まっています。
PMDAは、医薬品の開発から承認審査、再審査に至るまで、レジストリデータなどのリアルワールドデータの活用に向けて取り組んでいます。
2017年には条件付き早期承認制度(※)が法制化され、承認条件として実施を求める調査に、リアルワールドデータを活用した調査を含めています。
※条件付き早期承認制度:重篤な疾患で有効な治療方法が乏しく患者数が少ない疾患などを対象とする医薬品について、国内での治験実施が困難な場合や実施可能であってもかなりの長期間を要する場合には、検証的臨床試験(フェーズⅢ)の成績を求めることなく、市販後に必要な調査などを実施することを条件として製造販売承認を行う制度。
日本でのリアルワールドデータ活用に向けた取り組み
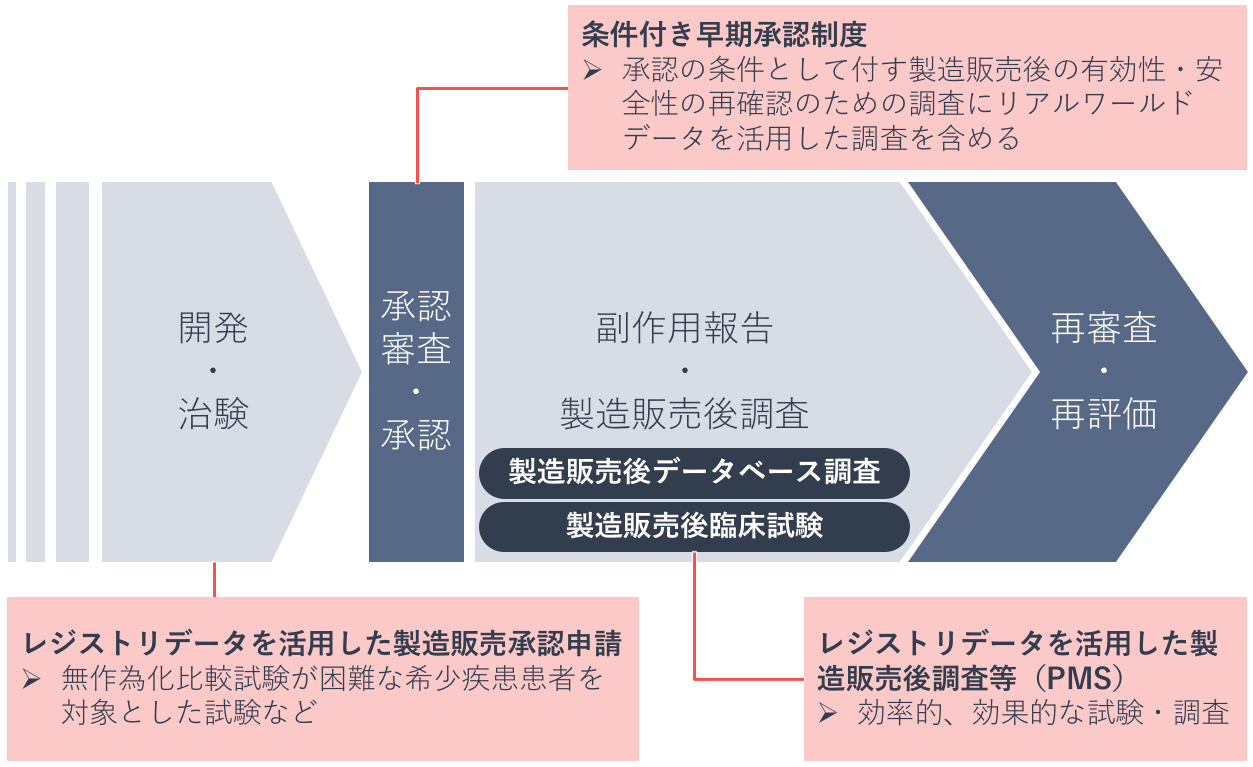
PMDA資料より改変作成
GPSPの目的を思い出す
最後に、GPSPの目的を思い出しましょう。GPSPの目的を理解し、自社医薬品の有効性と安全性の確保のためにMRがすべきことを改めて確認しましょう。
安全対策の背景に薬害あり
医薬品の安全対策の歴史は、繰り返されてきた薬害の歴史であるとも言えます。表に示したように、これまで国は、薬害が発生するたび、その教訓から安全対策を強化してきました。今では信じられないことに、かつては、医薬品の市販後に何らかの調査を行うという制度はなかったのです。薬害が繰り返され、市販後の安全性情報がいかに大切かが認識されるようになり、実臨床下の安全対策が制度としてスタートします。医薬品の再審査・再評価制度が導入されたのは、1979年の薬事法(※)改正からです。
※薬事法:現医薬品医療機器等法。2014年の改正時に現在の名称に改題された。
これまでの薬害と安全対策措置
| 原因(薬剤名等) | 報告・発生年 | 発生内容 | 発生後の措置等 |
|---|---|---|---|
| サリドマイド | 昭和36年 | 四肢奇形等 | 昭和38年から催奇形性試験 昭和42年医薬品の承認等に関する基本方針 |
| キノホルム | 昭和30年代 | スモン 神経炎症状 下半身麻痺 |
昭和54年医薬品副作用被害救済基金設立 薬事法改正:再審査・再評価制度新設 |
| 血液凝固第Ⅷ因子・第Ⅸ因子製剤 | 昭和60年代 | AIDS | 平成8年薬事法改正:感染症報告が義務化 |
| 輸血・血漿分画製剤 | – | B型肝炎・C型肝炎 | |
| ソリブジンと5-FUの相互作用 | 平成5年 | 重篤な血液障害 | 平成6年薬事法施行規則改正:未知・重篤30日→15日 平成8年薬事法改正:添付文書記載要領変更 |
| ヒト乾燥硬膜 | 平成8年頃から発生率上昇 | CJD | 平成14年薬事法改正:生物由来製品に関する規則、感染症定期報告 |
株式会社SCICUS「MR育薬学」より改変作成
製造販売後調査に新たに製造販売後データベース調査が規定された背景にもまた、残念なことに薬害がありました。
2008年にフィブリノゲン製剤によるC型肝炎薬害の全国原告団・全国弁護団と厚生労働大臣との基本合意書が交わされ、同年より「薬害肝炎事件の検証及び再発防止のための医薬品行政のあり方検討委員会」が開催されました。約2年にわたる検討の結果、最終提言(※)が取りまとめられ、この中で市販後安全対策として、電子レセプトなどのデータベースを活用した医薬品安全対策措置の効果評価のために情報基盤を整備する必要性が唱えられました。ここからリアルワールドデータの安全対策への活用方策についての議論が始まったのです。
つまり市販後安全対策(PMS)の究極の目的は、薬害の再発防止なのです。
※最終提言:平成22年4月 薬害肝炎事件の検証及び再発防止のための医薬品行政のあり方検討委員会「薬害再発防止のための医薬品行政等の見直しについて(最終提言)」
情報「収集」もMRの職務
製造販売後調査を行う際の製薬企業内の関係部門とその役割、MRの役割を改めて整理しておきましょう。
製造販売後の調査・試験に関する製薬企業内の実務と役割
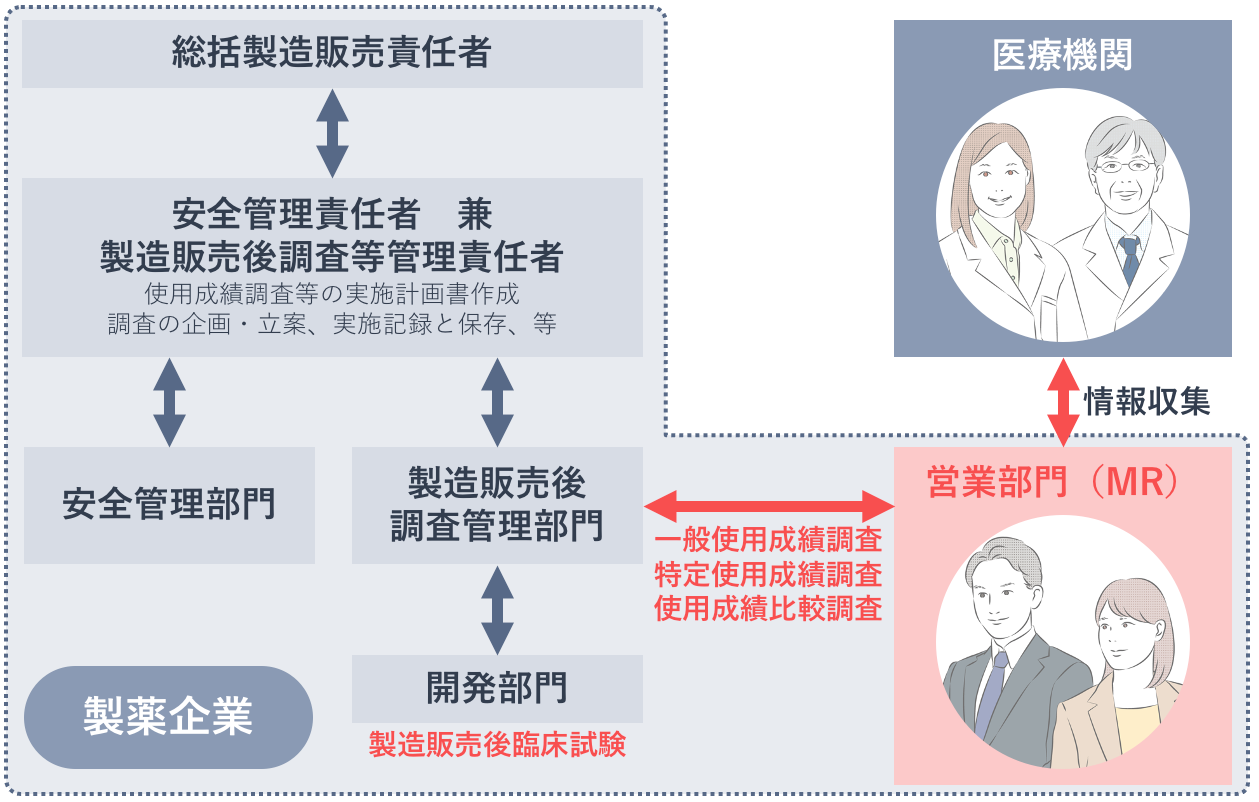
医薬品食品衛生研究所資料より改変作成
MRの皆さんの実務としては、一般使用成績調査や特定使用成績調査に協力してくれる医療機関への依頼、契約、調査票の回収があります。しかし最近では、製造販売後調査に関する業務は製造販売後調査管理部門の仕事であり、CRO(※)などに任せることが当然のことと考えるMRが増えているように感じます。
しかし、MRの職務を思い出してください。MRとは「医薬品の適正な使用に資するために、医療関係者を訪問すること等により安全管理情報を収集し、提供することを主な業務として行う者」(GVP省令第2条5)です。つまり、医薬品の有効性・安全性に関する情報を「収集」する製造販売後調査に関する業務もまた、MRの職務なのです。情報「提供・伝達」だけを行うMRは、MRの職務を果たしていないと言えます。
製造販売後調査に新たにデータベース調査が加わりましたが、製造販売後調査のすべてがデータベース調査で完結するわけではなく、引き続きMRが医療機関から収集する情報は欠かせません。とかく適切な情報「提供」が注目されがちなMRですが、適切な情報「収集」ができているかどうか、常に自身の行動を顧みるようにしましょう。
なお、過去に不適切事例としてPMDAから指摘、厚生労働省から指導が入った事案がありましたが、担当医師が記入すべき調査票をMRが代筆する行為は、たとえ内容の改ざんなどを行っていなくとも、データの信頼性を損なうことになるため、絶対にしてはならない行為です。
※CRO(Contract Research Organization):受託臨床試験実施機関のこと。製造販売後調査の業務も受託することがある。
MRの皆さんへ
臨床開発職の立場から、MRの皆さんへメッセージがあります。
臨床開発職にとって新薬は、時間をかけ大事にしてきた子どものような存在です。これはすべての臨床開発職が持っている気持ちです。社会に出た我が子をいつまでも見守り続け、その成長が一番の喜びとなります。
つまり製造販売承認がおり、上市された後も、心配なのです。もしかしたら治験で発見できなかった重大な有害事象が起こるかもしれない。それによる販売中止や売上ダウンといった会社の損害ももちろん心配ですが、何より、患者さんが苦しむことにならないかが心配なのです。
臨床開発職は、製造販売後の有害事象に関わる自発報告や安全性定期報告で何か問題が起こっていないか、気にかけています。また一般使用成績調査では、予定症例数に向けて契約は進んでいるのか、何例回収されているのか、その内容は信頼性のあるものか、不安に感じています。当然、医師たちが満足する有用性も気になります。会社を代表して定期的に医療機関に訪問するMRからの情報を待っているのです。
せっかく上市した薬を、10年先、20年先まで医療現場で活躍させるためにはMRの力が必要です。MRの皆さんも、医療に貢献したいという気持ちを発揮して、臨床開発職とともに社員全員で、自社医薬品を我が子のように育んでほしいと期待しています。

- 臨床開発職にとって新薬は、時間をかけ大事にしてきた子どものような存在です。
- 製造販売承認がおり、上市された後も、治験で発見できなかった重大な有害事象が起こらないか、患者さんが苦しむことにならないか心配です。
- 一般使用成績調査では、予定症例数に向けて契約は進んでいるのか、何例回収されているのか、その内容は信頼性のあるものか不安に感じており、会社を代表して定期的に医療機関に訪問するMRからの情報を待っています。
- MRの皆さんも、自社医薬品を我が子のように育んでほしいと期待しています。
医薬品は人間の健康を守るために欠かせないものです。しかし時に、有害事象を起こし人を傷つけることがあります。
MRは、安全性・有効性に関する情報を収集することで、医薬品のベネフィット/リスク比を最大化し、患者さんならびに国民、社会に貢献することが期待されています。それがMRの皆さんの使命であり、モチベーションでもあるでしょう。
市場に出ているすべての自社医薬品について、臨床現場での使用経験に基づく情報を収集すること、そして、改良や次の創薬に向けて開発の現場にフィードバックしていくこと。このMRの責務を、常に心に留めておきましょう。
MRの皆さんへ
- MRは、安全性・有効性に関する情報を収集することで、医薬品のベネフィット/リスク比を最大化し、患者さんならびに国民、社会に貢献することが期待されている
- 市場に出ているすべての自社医薬品について、臨床現場での使用経験に基づく情報を収集すること、改良や次の創薬に向けて開発の現場にフィードバックしていくことがMRの責務である
出典
- 文部科学省「平成24年度大学間連携共同教育推進事業」選定取組 四国の全薬学部の連携・共同による薬学教育改革 2014年7月11日講演会「医薬品の製造販売後の安全性確保に関する行政施策と、関連するバイオマーカーおよび薬剤疫学研究」資料
- 厚生労働省令第155号
- 2018年10月18日 平成30年度第7回医薬品医療機器制度部会資料1
- 2017年3月30日 第1回医薬品医療機器制度部会資料4
- 2018年5月9日 第2回医薬品医療機器制度部会資料1-1
- 医薬品の製造販売後の調査及び試験の実施の基準に関する省令
- 2018年2月21日付 薬生薬審発0221第1号厚生労働省医薬・生活衛生局医薬品審査管理課長通知
- 2013年12月18日 第1回医療情報データベース基盤整備事業のあり方に関する検討会資料2
- 2019年3月13日 第5回臨床開発環境整備推進会議資料1-3
- PMDAウェブサイト(2021年3月アクセス)
- 2019年6月22日 第27回抗悪性腫瘍薬開発フォーラム資料
- 2020年11月13日 PMDA令和2年度第2回運営評議会資料3
- メディカルエデュケーション編集部編 (2017年) MR育薬学 株式会社SCICUS
- 医薬品、医薬部外品、化粧品、医療機器及び再生医療等製品の製造販売後安全管理の基準に関する省令